Qu’est-ce que c’est ?
La maladie de Creutzfeldt-Jakob fait partie des maladies à prions. Ce sont des maladies rares caractérisées par une dégénérescence du système nerveux central et qui sont également appelées encéphalopathies subaiguës spongiformes transmissibles (ESST).
Elles sont dues à l’accumulation dans le cerveau d’une protéine normale mais mal conformée, la protéine prion(1). Malheureusement, la maladie de Creutzfeldt-Jakob se caractérise par une évolution rapide et fatale ainsi que par l’absence de traitement. On compte chaque année 100 à 150 cas en France(2).
Les symptômes
La maladie débute souvent par des troubles non spécifiques comme l’insomnie ou l’anxiété. Peu à peu, des troubles de la mémoire, de l’orientation et du langage s’installent. Elle se manifeste ensuite par des troubles psychiatriques ainsi qu’une ataxie cérébelleuse (instabilité en station debout immobile et pendant la marche qui s’accompagne d’une titubation analogue à celle de l’ivresse). On note également des lésions typiques dans le système nerveux central (plaques florides, dépôts amyloïdes de PrPres entourés de vacuoles).
Les deux sexes sont atteints avec toutefois une fréquence élevée chez les adultes jeunes.
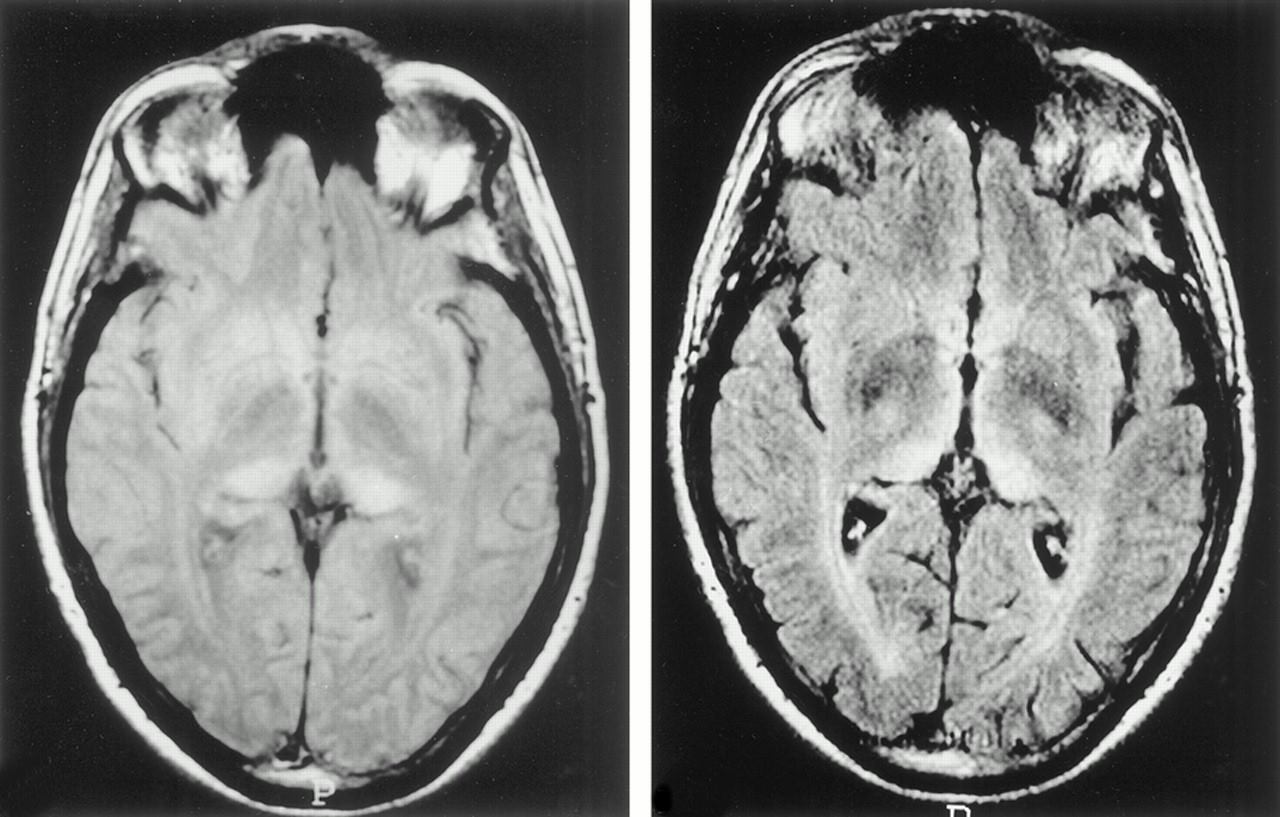
Il n’existe malheureusement aucun test diagnostic fiable. Un électroencéphalogramme (EEG) permet de repérer des perturbations relativement spécifiques de l’activité cérébrale. L’IRM met en évidence des anomalies particulières au niveau de certaines régions du cerveau (noyaux gris centraux, cortex) pour lesquelles les diagnostics différentiels sont peu nombreux.
Si l’ensemble de ces éléments cliniques et paracliniques peut permettre de poser un diagnostic de la maladie de Creutzfeldt-Jakob, il ne s’agit que d’un diagnostic de probabilité : en effet, seul l’examen du tissu cérébral, réalisé le plus souvent après la mort, permet d’affirmer le diagnostic.
Les origines de la maladie
La maladie de Creutzfeld-Jakob est la seule maladie humaine à pouvoir être de cause génétique (due à une mutation dans le gène codant pour la protéine prion, la mutation E200K étant la plus fréquente), de cause infectieuse (secondaire à une contamination) ou de forme sporadique (de survenue aléatoire, sans mutation ni exposition à un prion exogène retrouvée).
Toutefois, la forme sporadique est la plus fréquente : elle représente 85 % de l’ensemble des encéphalopathies subaiguës spongiformes transmissibles (ESST) diagnostiquées chaque année. Dans ce cas, la maladie apparaît généralement après 60 ans et évolue sur une période d’environ 6 mois. Lorsque la maladie est d’origine génétique ou infectieuse, les symptômes sont plus précoces et d’évolution plus lente. Dans les formes infectieuses, la période d’incubation peut être extrêmement longue et dépasser 50 ans.
Les facteurs de risque
La protéine prion (PrPc) est une protéine physiologique qu’on retrouve de façon très conservée chez de nombreuses espèces. Dans les neurones du cerveau, la protéine prion peut devenir pathogène en changeant sa conformation tridimensionnelle : elle se replie sur elle-même de façon très serrée, ce qui la rend hydrophobe, peu soluble et résistante à la dégradation.
On l’appelle alors protéine prion « scrapie » (PrPsc). Les PrPsc s’agrègent entre elles et forment des dépôts qui se multiplient à l’intérieur et à l’extérieur des cellules du cerveau, perturbant leur fonctionnement et leurs mécanismes de survie.
Dans cette forme anormale, la protéine prion est en outre capable de transmettre son anomalie conformationnelle : au contact d’une PrPsc, une protéine prion normale adopte à son tour une conformation anormale. C’est l’effet domino.
Le risque de transmission entre individus
La transmission interindividuelle des maladies à prions est possible en cas de greffe de tissus ou suite à l’administration d’hormones de croissance. Les tissus plus à risque sont issus du système nerveux central et de l’œil. Dans une moindre mesure, le liquide céphalorachidien, le sang et certains organes (reins, poumons…) peuvent aussi transmettre le prion anormal.
Le risque lié à l’alimentation
La transmission d’un prion du bovin à l’Homme via la consommation d’aliments contaminés est suspectée en 1996, lors de la dramatique crise de la « vache folle ». Depuis plusieurs années, une épidémie d’encéphalopathie spongiforme bovine (ESB) frappait les élevages du Royaume-Uni3. La propagation de cette maladie à prions, qui touchait plusieurs dizaines de milliers de bêtes chaque année, était sans doute due à l’utilisation de farines animales, produites à partir de carcasses et insuffisamment décontaminées. Son origine reste toutefois discutée.
Prévention et traitement
Aujourd’hui, il n’existe pas de traitement spécifique des maladies à prions. Les seuls médicaments qui peuvent être prescrits sont ceux qui peuvent soulager ou limiter les différents symptômes de la maladie. Une aide à la prise en charge médico-sociale et psychologique est proposée aux malades et à leur famille par la Cellule nationale d’aide à la prise en charge des MCJ. La recherche de médicaments visant à empêcher la conversion de la PrPc, favoriser l’élimination des formes anormales de la protéine et en limiter la propagation est porteuse d’espoir.
Une piste intéressante cible PDK1, un des médiateurs cellulaires mis en jeu au cours de l’infection. Son inhibition permettrait à la fois d’inhiber le phénomène de conversion en favorisant le clivage de la PrPc, et d’atténuer les conséquences de sa réplication sur la survie des neurones.
Références bibliographiques
Notes:
1. Prusiner S, Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012 Jun 22; 336(6088):1511-3.
2. Diack AB et al. Variant CJD. 18 years of research and surveillance. Prion. 2014; 8(4):286-95.
3. Barria MA et al.Exploring the zoonotic potential of animal prion diseases: in vivo and in vitro approaches. Prion. 2014 Jan-Feb; 8(1):85-91
Références
Dormont D. Les encéphalopathies subaiguës spongiformes transmissibles ou maladies à prions. Méd. Mal. Infect., 2001, 31, suppl 2, 288-97.
Dubreuil G.,Contamin H., Dormont D. Prions et primates non humains. Primatologie, 2001, 4, 373-389. – Baron T. Transmissibilité de l’encéphalopathie spongiforme bovine à l’homme : état des connaissances actuelles . Point vét., 2000, 31, 184-89.
Baron T : De la vache folle à l’homme. Ed Michel Servent, Lyon 2002.
Laisser un commentaire